STEP-HFpEF: Semaglutide in patients with HF & ejection fraction >=45% & obesity
STEP-HFpEF main paper. N Engl J Med. 2023;389:1069-84.
STEP-HFpEF sub-analysis by baseline BMI/achieved weight loss. Nat Med 2023;2358-65
STEP-HFpEF subgroup by baseline LVEF category. JACC 2023;82:2087-96
Bottom line: In patients with symptomatic HF, ejection fraction >=45% & BMI >=30, semaglutide improved quality of life & reduced weight (average ~11% weight loss at 1 year), but increased the risk of discontinuation due to GI intolerance. For every 100 patients treated for 1 year, 12 patients will get a noticeable improvement in their quality of life because of semaglutide, & 8 patients will discontinue it due to intolerable side-effects (mostly gastrointestinal).
Patients (n=529 randomized)
13 countries, 2021-2022
Included:
Age >=18 years
Symptomatic (NYHA 2-4) HF with left ventricular ejection fraction >=45%
At least one of the following:
Elevated LV filling pressures;
Elevated BNP/NTproBNP plus echocardiographic abnormalities;
HF hospitalization in the last 12 months, plus ongoing treatment with diuretics OR echo abnormalities
BMI >=30
Kansas City Cardiomyopathy Questionnaire (KCCQ)-Clinical Summary Score (CSS) <90/100
6-minute walk distance (6MWD) >=100 meters
Key exclusions:
Body-weight change >5 kg in last 90 days
Diabetes: Prior diagnosis, A1c >=6.5% at screening
ESRD or dialysis dependence
Baseline:
Age 69 y, male 44%
White 96%, Black 4%
LVEF median 57% (45-49% in 16%, >=60% in 43%)
NYHA 2 66%, 3-4 34%
KCCQ-CSS median 59/100, 6MWD median 320 meters
HF hospitalization in last year 15%
Weight median 105 kg, BMI median 37 (66% BMI >=35)
Comorbidities: AF 52%, CAD 18.5%, HTN 82%
Meds: Loop diuretic 62%, ACE/ARB/ARNI 80%, beta-blocker 79%, MRA 35%, SGLT2i 4%
Intervention: Semaglutide subcutaneous once weekly at “weight loss doses”
Starting dose: 0.25 mg q1w
Titration: Uptitrated every 4 weeks (to 0.5 -> 1.0 -> 1.7 -> 2.4 mg q1w) as tolerated
Target dose: 2.4 mg q1w (reached after 16 weeks)
84% of those still taking the drug at 1 year received the target dose
Comparator: Matching placebo
Outcomes @ 1 year
Co-primary outcomes: Mean change from baseline to week 52:
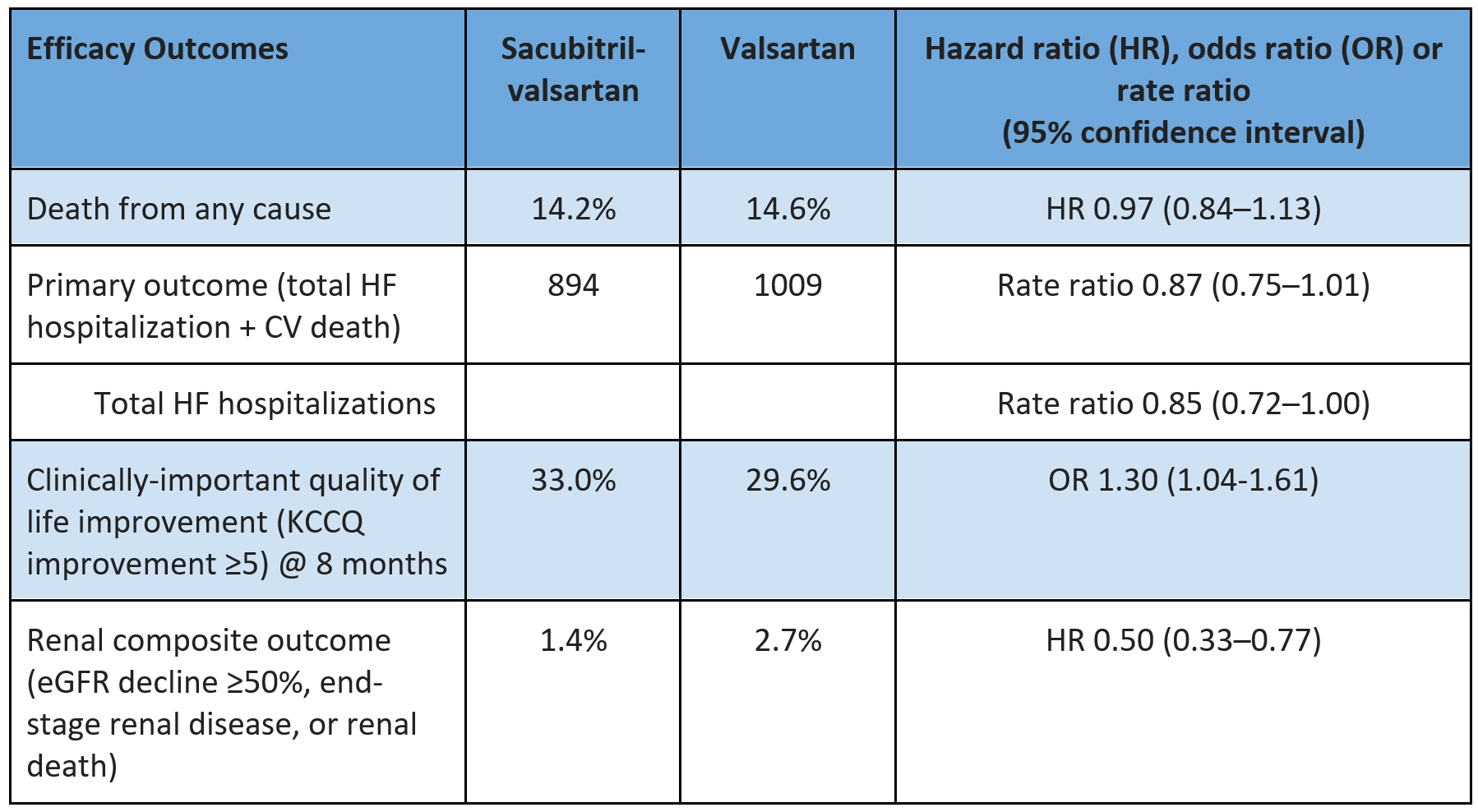
KCCQ-CSS: Semaglutide +16.6/100 vs placebo +8.7/100 - mean difference +7.8
Clinically-important improvement (>=5-point improvement) in KCCQ-CSS: 75.3% vs 63.7% (+11.6%)
Large improvement (>=20-point improvement) in KCCQ-CSS: 37.9% vs 26.6% (+11.3%)
Consistent mean improvement over placebo across KCCQ-Overall Summary Score (+7.5), across sub-scores (e.g. Total Symptoms Score +9.0) & individual domains, including social limitations score (+6.7)
Weight: Semaglutide -13.3% vs placebo -2.6% - mean difference -10.7%
>=20% reduction: 23.6% vs 0.4%
Key secondary outcomes
6MWD: mean +20 meters with semaglutide vs placebo
Exploratory HF composite (HF hospitalization or urgent visit): 0.3% vs 4.5% (hazard ration 0.08, 95% confidence interval 0.00-0.42)
Safety
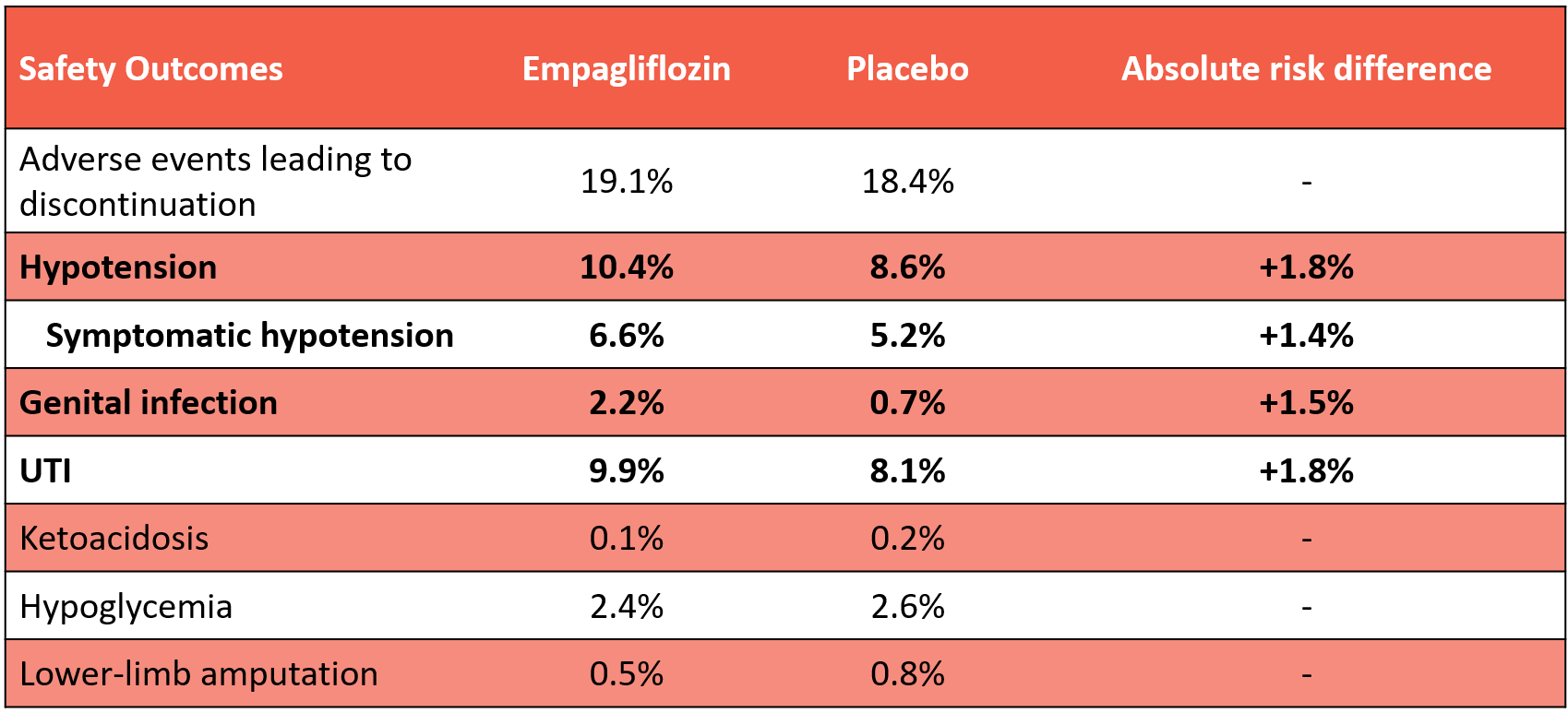
Serious adverse events: Semaglutide 15.2% vs placebo 28.2%
Discontinued due to adverse events: 13.3% vs 5.3%
Internal validity = low risk of bias
Computer-generated random sequence generation
Allocation concealment by centralized interactive web-based response system
Blinding by matching placebo & titration schedule
Intention-to-treat analysis
Loss to follow-up (LTFU): KCCQ data missing for 8% on semaglutide & 11% on placebo at 1 year
Generalizability & other considerations
Similar improvement (no significant treatment-subgroup interaction) in QoL with semaglutide across studied LVEF in mildly-reduced/preserved range (45% to >=60%)
Similar improvement in QoL with semaglutide regardless of BMI (but all >=30), but KCCQ-CSS improvement in the semaglutide group was associated with weight loss >=5%
Impossible to say whether lesser KCCQ improvement in patients who lost <5% of their body weight due to an actual cause-effect relationship between weight loss & QoL improvement, or whether this is confounded by some other factor (e.g. lower adherence to semaglutide could explain lack of both weight loss & QoL improvement)